
Abstract
Malignant transformation of peripheral nerve sheath tumor (MPNST) may develop from a plexiform type of Neurofibromatosis 1 (NF1) or previously irradiated areas. Generally, MPNSTs occur in about 2% to 5% of neurofibromatosis patients. In this paper, we present a 58-year-old male patient with neurofibromatosis who developed MPNST of the eyelids and nasal area. The patient had a history of multiple excision biopsies for facial tumors in 22 years at different institutions, allegedly revealing neurofibromas on histopathological evaluation. A recent consult with the Otorhinolaryngology Service (ORL) prompted an excision biopsy with results consistent with neurofibroma. The mass recurred and enlarged even more rapidly compared to the previously excised tumor. The patient also developed four tumors on the eyelids hence the referral to Ophthalmology Service. The eyelid masses and nasal mass were excised by the Ophthalmology and ORL Services. Histopathology revealed identical MPNST characteristics on all excised tumors. The patient was eventually referred to the Oncology Service to evaluate radio and chemotherapy. A rapid change in the size of a preexisting neurofibroma, infiltration of the adjacent structures, intralesional hemorrhage, and pain usually indicates a possible malignant transformation into MPNST. A high index of suspicion is helpful for clinicians when presented with a case of a recurrent neurofibromatosis, even if the only sign is the rapid growth of the mass since management of MPNST is very different from neurofibromatosis.
Author Contributions
Academic Editor: Chen R, United States
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2021 Jib Alabado, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
History and PE
A 58-year-old Filipino male presented with multiple, soft, painless masses on the forehead and scalp region for 22 years. There was no blurring of vision, diplopia, or other associated symptoms reported by the patient. In the interval, there was noted the recurrence of masses over the scalp region. Previously in the same area, excisions and recurrences were done in 2004, 2005, 2011, and 2016. Results of histopathology allegedly showed neurofibromatosis. One year earlier, there was a note of recurrence of soft tissue masses on bilateral upper and lower lids, rapidly increasing in size and with extension to the right nares. The gradual obstruction of the right nostril with exophytic, fleshy mass with irregular borders prompted consult with ENT service. A right nasal biopsy was then done, which showed a recurrence of neurofibromatosis. Furthermore, the bilateral upper and lower lid masses rapidly increased in size hence the referral to Ophthalmology Service. The patient noted that aside from the rapid growth rate, the lesions were very similar to the previous tumors – firm, movable, and non-tender.
On ophthalmologic examination, the best-corrected visual acuity (BCVA) for the right eye was 6/7.5 and 6/6 for the left eye. There was no proptosis on Hertel’s exophthalmometry. On the right upper eyelid, a 3x3x1.8 cm erythematous firm mass with telangiectatic vessels was visualized and palpated on the nasal area and a 1.1x0.9x1.0cm non-erythematous mass, firm, non-tender, mass was noted on the temporal area. The medial posterior lamella of the right lower lid appeared to be thickened. On the left upper eyelid, there was a 1.5x1.5cm soft, non-tender, nodular mass. A 1.7x1.5cm nodular, firm, rubbery mass was also noted on the medial aspect of the left lower eyelid. Eye motility was normal, and pupils were symmetric with no afferent pupillary defect. Intraocular pressures were normal, no abnormalities on anterior and posterior segment examination. On palpation, there was a left submandibular lymphadenopathy. Facial, Cranial, and Orbital MRI noted an 8.6 x 7.3 x 6.0 cm (CCxWxAP) lo bulated, fungating mass arising from the right nasal region exhibiting isointense signals on T1-W and hypertintense to muscle in T2-W. The mass showed avid enhancement on postcontrast scans. The mass extended into the right nasal cavity and was intimately related to the inferior turbinate. Posteriorly, the mass appeared to involve the medial aspect of the right maxillary bone. A lobulated,fungating mass arose from the medial right intraorbital extraconal region measuring approximately 3.2 x 3.3 x 4.5 cm. This was also isointense on TW1, and hyperintense to muscle on T2W similar to the nasal mass and intimately related to the medial aspect of the globe and medial rectus muscle. The orbicularis oculi muscle on the right was not delineated. Medially, the mass was bounded by the lamina papyracea. Multiple smaller similar-looking subcutaneous foci are Figure 1. Preoperative picture showing the extent of lesions. (A) 1.0x0.9x1.0cm firm, non-tender mass (B) 3.0x3.0x1.8cm soft, erythematous, telangiectatic mass (C) 1.5x1.5cm soft, non-tender, nodular mass (D) 1.7x1.5cm firm, rubbery, nodular mass (E) 8.6x7.3x6.0cm firm, fungating nasal mass excised by ORL service.
Figure 1.Preoperative picture showing extent of lesions. (A) 1.0x0.9x1.0cm firm, non-tender mass (B)3.0x3.0x1.8cm soft, erythematous, telangiectatic mass (C) 1.5x1.5cm soft, non-tender, nodular mass (D) 1.7x1.5cm firm, rubbery, nodular mass (E) 8.6x7.3x6.0cm firm, fungating nasal mass excised by Otorhinolaryngology service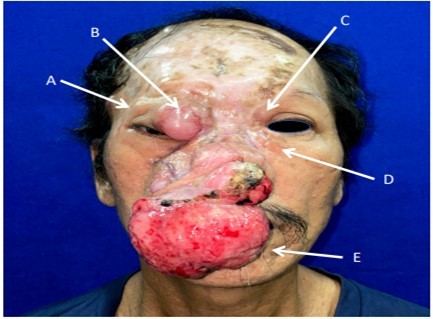
There was a similar focus at the subcutaneous layer at the right temporal region, measuring 1.8 x 0.9 x 1.7 cm. Skin thickening and subcutaneous edema at the right lateral periorbital region were noted with increased contrast enhancement. The radiologic impression was of multiple lesions in the face and right temporal cutaneous area, likely neurofibromas. At this point in the patient’s history and physical examination, we had a lesion consistent with the patient’s previous lesions, diagnosed as neurofibromas. This was further supported by radiologic evidence of lesions similar to neurofibromas. The only inconsistency was the rapid growth rate not normally seen in the patient’s previous lesions and contradictory to the slow, indolent growth exhibited by typical neurofibromas.
Differential Diagnosis
With the previous biopsy done 5 months ago, the primary working impression is neurofibromatosis. These types of lesions tend to be painless, slowly growing, solitary, skin-colored, soft, flaccid, rubbery to firm papules or nodules with a smooth surface measuring up to 2 cm. Classic characteristics include a “bag-of-worms” consistency and eyelid involvement producing an S-shaped deformity in cases of the plexiform variants1. The lesion also invaginates with pressure. The prevailing diagnostic criteria of Figure 2. Multiple facial lesions are radiologically consistent with neurofibromas. (A) 1.8x0.9x1.7cm right lateral temporal mass (B) 3.2x3.3x4.5cm right medial intraorbital mass (C) 8.6x7.3x6.0cm right nasal mass neurofibromatosis type 1 are met if 2 or more of the following are present: (1) ≥ 6 café au lait patches (2) ≥ 2 neurofibromas of any type, or 1 plexiform neurofibroma (3) axillary or inguinal freckling (4) ≥ 2 lisch nodules, (5) optic glioma, (6) sphenoid dysplasia or thinning of long bone cortex with or without pseudoarthrosis (7) first-degree relative diagnosed with neurofibromatosis type12. In contrast to this, our patient did not satisfy any clinical manifestations of Neurofibromatosis type 1. The closest differential of neurofibroma would be a schwannoma. If present on the orbit, they are insidious and proptose gradually without pulsations. Most tumors infiltrate the superior quadrant, causing inferiorly displaced proptosis or frank hypoglobus3. Patients may experience diplopia, eye movement limitation, diminished visual acuity, and optic nerve compression symptoms, including scotomas dyschromatopsia, and impaired contrast sensitivity. They may also experience pain or paresthesia in the distribution of the nerve. In severe cases, there may be a palpable orbital mass4. Only one case has been reported of a bilateral orbit involvement5. Another differential would be the malignant transformation of neurofibroma. Malignant peripheral nerve sheath tumor may arise from a preexisting nerve sheath tumor in neurofibromatosis type 1 and can arise in virtually any anatomic location. The most common sites are the trunk and extremities, followed by the head and neck. There is no gender preference, and it tends to occur in younger individuals with Neurofibromatosis1. Histologic evaluation is necessary but not always specific and requires correlation with clinical and radiologic findings6. At this point, the clinical presentation is consistent with the previous histopathologic diagnoses, that of a neurofibroma. Although a neurofibrosarcoma would also have a similar presentation at its early stages, its predilection for arising from NF type 1 lesions is not seen in our patient.
Figure 2.Multiple facial lesions radiologically consistent with neurofibromas. (A) 1.8X0.9X1.7cm right lateral temporal mass (B) 3.2X3X4.5 cmright medial intraorbital mass (B) (C) 8.6X7.3X6.0 cmr right nasal mass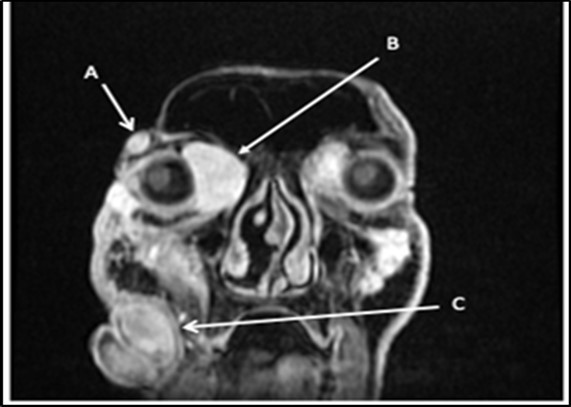
Surgical Management
multidisciplinary discussion with Ophthalmology and ENT service was conducted to plan the surgical approach. The combined surgical management was “Excision biopsy with eyelid reconstruction, possibly skin graft for reconstruction of the anterior lamella from the supraclavicular area, with Excision of Right intranasal mass with frozen section and reconstruction using an anterolateral thigh flap under general anesthesia”. If the skin is most likely too thin and cannot be used to cover the defect after excision, the plan was to harvest a skin graft from the supraclavicular area. Upon injection of local anesthesia subcutaneously, it was noted that the skin was easily dissected from the mass, and the vessels blanched. The skin was assessed to be thicker and of better quality, than expected. The loss of the telangiectatic vessels meant that hemostasis may be achieved and would not pose a bigger complication as expected. Intraoperatively it was decided to save the skin for closure instead of harvesting a graft. Extensive dissection and mobilization were applied up to the extraconal extent of the right medial mass. After its excision, a large cavity was noted. The tumor on the right temporal area of the eyelid was next excised by extending the incision from the nasal area to the temporal area via lid crease approach. Tumors located on the medial aspect of the superior and inferior left eyelids were excised, imploring the same technique. Huge cavities were noted; hence extensive undermining of the subcutaneous tissue on the adjacent skin territories was initiated to avoid tension during closure. Intraoperatively the gross appearance of the excised lesions was consistent with a neurofibroma. We noted whitish-tan lesions with a glistening surface, appearing encapsulated, with diffuse soft tissue infiltration suggestive of neurofibromas. The lesions were also not vascular, which we would have expected in a malignancy. Likewise, not seen were signs of bony erosion or frank tumor necrosis. On post-op, the patient did not experience fever nor complain of any discomfort or pain. Wound dressing was not noted to be soaked, and no serosanguinous discharge was seen at the wound site.
Discussion
We have a case whose diagnosis needs to be confirmed. The previous biopsy showed histopathology consistent with a neurofibroma, with monomorphic, spindle-shaped, distinct cells, well-spaced, with scant mitosis. Neurofibromas are an autosomal dominant hereditary disease with a varied phenotype that affects the skin and nervous system. Affectation is one out of every 3,500 live births throughout the world. A spontaneous mutation on chromosome 17 is thought to be responsible for around half of the cases7. Figure 3. Comparison between biopsies taken five months apart. The histopathology in the biopsy done in February shows well-spaced uniform cells (black arrows). The biopsy done in July shows cellular congestion with atypical, pleomorphic cells with increased mitotic activity (blue arrows). In the most recent procedure, routine histopathology showed a different picture with higher mitotic figures and cellular congestion. The increased mitotic activity causes cellular production at a greater rate that the resulting cells show atypia and ill-defined borders. Hemorrhage and necrosis are also evident, hinting the tumor growth exceeds its vascular supply. The rapid clinical growth supported the high proliferative nature of the tumor. A diagnosis of Spindle cell sarcoma was made, with low to intermediate grade; however, a final diagnosis cannot be made based on morphology alone. To determine the type of malignancy, a panel of immunohistochemical stains was ordered. Table 1. Immunohistochemical Stain Marker S-100 Tumors of Mesenchymal origin (Sarcomas) SMA Myoepithelial cells Desmin Sarcoma vs Neurofibroma CD34 Epithelioid Sarcoma vs Dermatofibroma CK Tumors of Epithelial origin (Carcinomas) EMA Adenocarcinoma Three screening stains were ordered to identify the origin of the malignancy. S- 100 is a screening stain that becomes positive when exposed to mesenchymal cells. CK screens for malignancies of epithelial origin, while CD34 screens for hematopoietic involvement. Smooth Muscle Actin is a stain used to determine the presence of myoepithelial cells. This was chosen to consider the possibility of pulmonary metastasis. Desmin is a stain chosen due to its ability to differentiate rhabdomyosarcomas from other mesenchymal malignancies. Finally, EMA is a marker used to confirm carcinomatous lesions8 . Since the gross appearance of the tumor closely resembled muscular tissue, the S-100 stain was done first. The stain clearly showed a positive reaction. There was a note of mitotic activity in up to 7/10 high power fields with associated necrosis. This clinched the diagnosis as a mesenchymal tumor, allowing us to rule out three stains – CD34, CK, and EMA, along with their respective differentials of hematopoietic and epithelial tumors9. The initial positive reaction to S-100 allowed a narrowing of differentials to mesenchymal cell origin tumors. We could consider neurofibromas, schwannomas, malignant peripheral nerve sheath tumors, as well as metastatic myoepithelial cells. To help us differentiate, we take a closer look at SMA next. The sample did not stain to SMA, producing a negative result. This effectively ruled out myoepithelial tumors such as metastatic pleuropulmonary tumors, narrowing our differentials to neuronal tumors and smooth muscle rhabdomyosarcomas10. It was decided that determining desmin content would allow us to further narrow the differentials by differentiating between a rhabdomyosarcoma that would stain positively for desmin, and neuronal tumors such as a neurofibroma will be negative staining11,12 The specimen is composed of pleomorphic spindle cells forming sheets with a marbling pattern. Mitotic activity is present in up to 7/10 high power fields with associated crowding and necrosis. This portion of the lesion shows immunoreactivity for S100, while the remainder of the lesion has focal reactivity. Negative stains include SMA and Desmin. Since a neurofibroma can undergo malignant transformation, these last two lesions are what remain among our differentials. While morphologic histologic findings are more consistent with malignant growth, a closer look at the staining characteristics of S-100 can differentiate between the two lesions. In neurofibrosarcomas or malignant peripheral nerve sheath tumors, S100 staining is patchy and focal, with less than 50% of the specimen showing reactivity to staining. In neurofibromas, the S100 reaction is strong and diffuse, staining nearly the entire specimen13. The morphologic changes, immunohistochemical staining, and character of staining of S100 allows us to clinch the diagnosis of a Malignant Peripheral Nerve Sheath Tumor arising from a recurrent neurofibroma. Figure 4. Immunohistochemistry Staining results. S-100 shows selective staining of the cells of interest (black arrows). SMA and desmin showed negative staining of cells. In two population-based investigations, the lifetime probability of developing MPNST in NF1 patients was estimated to be 8-16 percent. Malignant transformation can begin as early as childhood, although it is most common in life's third to fourth decades. The probability of sarcoma-specific mortality is greatest in high-grade MPNST. Overall survival after five years ranges from 20% to 50%, with unresectable or metastatic cancer having a particularly poor prognosis14. Figure 5, Figure 6
Figure 3.Comparison between biopsies taken five months apart. The histopathology in the biopsy done in February shows well-spaced uniform cells (black arrows). The biopsy done in July shows cellular congestion with atypical, pleomorphic cells with increased mitotic activity (blue arrows).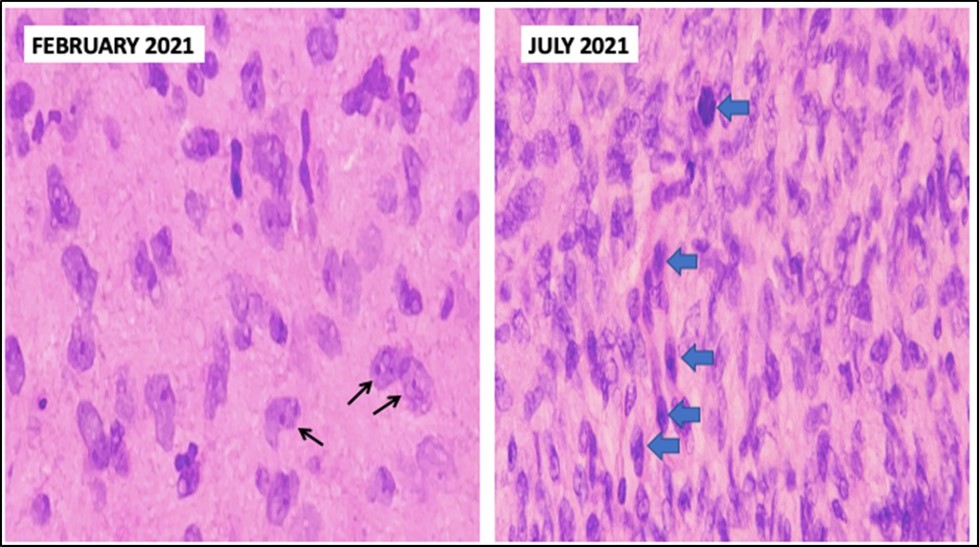
Figure 4.Immunohistochemistry staining results. S-100 shows selective staining of the cells of interest (black arrows).
Figure 5.Immunohistochemistry Staining results. Smooth Muscle Actin (SMA) showed negative staining of cells.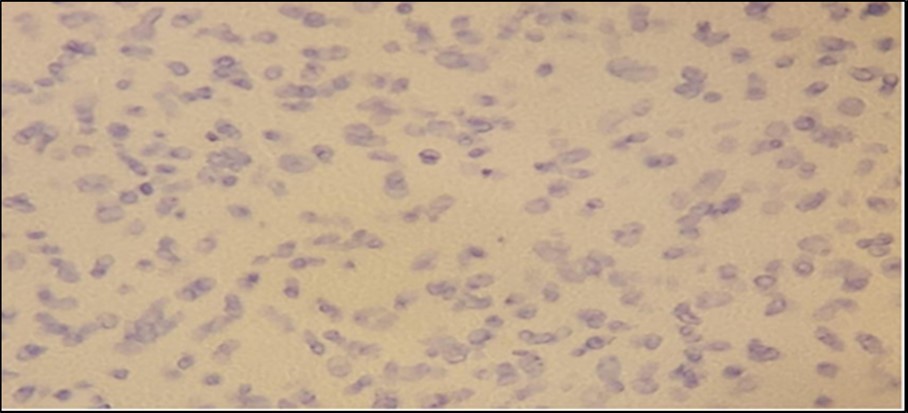
Figure 6.Immunohistochemistry Staining results. Desmin showed negative staining of cells.
In summary, the biopsy showed a histopathology of spindle cells. This specimen was subjected to the following stains: S100, desmin, and SMA. The profile derived from these stains concluded that the disease process is that of a Malignant Peripheral Peripheral Nerve Sheath Tumor.
Management
The prognosis of Malignant Peripheral Nerve Sheath Tumor is poor, especially for tumors that cannot be resected15 . A large tumor size, truncal location, and positive resection margins greatly predicted local recurrence, while tumor grade was significant for distant metastasis16. The mainstay of treatment is aggressive surgical resection with negative margins combined with adjuvant radiotherapy for large-sized, high-grade tumors with positive surgical margins with the option of additional chemotherapy with doxorubicin if the patient can tolerate the treatment17 . Currently, the patient is undergoing metastatic workup, the results of which should guide the direction of treatment and care and a more accurate prognosis.
References
- 1.Karcioglu Z. (2006) Clinicopathologic Correlates in Orbital Disease. Duane’s Foundations of Clinical Ophthalmology, Volume 3, Chapter 17. Lippincott Williams & Wilkins. edition on CD-ROM
- 2.Lee L R, etal. (2000) Localized neurofibroma of the orbit: a radiographic and histopathologic study.Ophthal Plast Reconstr Surg. 16(3), 241-6.
- 3.Singh M, etal. (2013) Clinico-Radiological Spectrum and Management of Orbital Schwannomas: A Tertiary Care Institute Study. Orbit [Internet].
- 4.Cantore G. (1986) Orbital Schwannomas: Report of Nine Cases and Review of the Literature. , Neurosurgery 19(4), 583-8.
- 5.Sales-Sanz M, etal. (2007) Bilateral simultaneous ancient schwannomas of the orbit. Ophthal Plast Reconstr Surg. Jan;23(1): 68-9.
- 6.RodriguezFJ etal.Genetic predisposition to peripheral nerve neoplasia: diagnostic criteria and pathogenesis of neurofibromatoses, Carney complex, and related syndromes. , Acta Neuropathol.2012 123(3), 349-67.
- 7.Blessmann M, etal.Adhesion molecule L1 is down-regulated in malignant peripheral nerve sheath tumors versus benign neurofibromatosis type 1–associated tumors. 113(2), 239-244.
- 10.Perez-Montiel M D. (2006) Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. , Am J Dermatopathol 28(2), 105-11.
- 12.Kresak J, etal. (2016) Neurofibromatosis: A Review of NF1, NF2, ans Schwannomatosis. , Journal of Pediatric Genetics
- 13.Agaram N P, etal. (2005) Deep-seated plexiform schwannoma: a pathologic study of 16 cases and comparative analysis with the superficial variety” Am J Surg Pathol. 29(8), 1042-8.
- 14.Miettinen M M, etal.Histopathologic Evaluation of Atypical Neurofibromatous Tumors and Their Transformation into Malignant Peripheral Nerve Sheath Tumor in Neurofibromatosis 1 Patients – A Consensus Overview. Human Pathology.
- 15.Samer A A, etal. (2020) Fungating malignant peripheral nerve sheath tumor arising from a slow-growing mass in the forearm: a case report and review of literature. , Journal of Medical Case Reports 14-91.